The intestinal epithelial lining, together with factors secreted from it, forms a barrier that separates the host from the environment. In pathologic conditions, the permeability of the epithelial lining may be compromised allowing the passage of toxins, antigens, and bacteria in the lumen to enter the blood stream creating a "leaky gut." In individuals with a genetic predisposition, a leaky gut may allow environmental factors to enter the body and trigger the initiation and development of autoimmune disease. Growing evidence shows that the gut microbiota is important in supporting the epithelial barrier and therefore plays a key role in the regulation of environmental factors that enter the body. Several recent reports have shown that probiotics can reverse the leaky gut by enhancing the production of tight junction proteins; however, additional and longer term studies are still required. Conversely, pathogenic bacteria that can facilitate a leaky gut and induce autoimmune symptoms can be ameliorated with the use of antibiotic treatment. Therefore, it is hypothesized that modulating the gut microbiota can serve as a potential method for regulating intestinal permeability and may help to alter the course of autoimmune diseases in susceptible individuals.
Introduction
For digestion and absorption purposes, mammals have developed a very complicated and highly specialized gastrointestinal system maintained by the mucosal barrier (1). However, apart from absorbable nutrients, the intestinal mucosa also faces tremendous exterior antigens, including food antigens, commensal bacteria, pathogens, and toxins. Thus, a specialized barrier function is required to block the entry of diverse exterior antigens while absorbing nutrients. Impressively, in the intestine, the front line of this barrier is maintained by only a single layer of specialized epithelial cells that are linked together by tight junction (TJ) proteins. Many other factors aid in support of this barrier including mucins, antimicrobial molecules, immunoglobulins, and cytokines. If any abnormalities occur among these factors, the intestinal permeability may increase, which is termed a "leaky gut." A leaky gut allows the entry of exterior antigens from the gut lumen into the host, which may promote both local and systemic immune responses. Multiple diseases may arise or be exacerbated due to a leaky gut, including autoimmune diseases such as inflammatory bowel disease, celiac disease, autoimmune hepatitis, type 1 diabetes (T1D), multiple sclerosis, and systemic lupus erythematosus (SLE) (2–6). Numerous factors can affect gut permeability, such as various diet-derived compounds, alcohol consumption, and gut microbiota dysbiosis. While this review is focused on chronic inflammation and gut barrier functions in mammals, it is worth noting that leaky gut is a phenomenon that is widespread in both mammalian and non-mammalian animals (7). Thus, studies in systems outside of mammals, such as zebrafish (7, 8), can be also helpful in our understanding of the relationship between inflammation and the intestinal barrier.
The gut microbiota has drawn intense attention in the past decade (9). Although scientists have studied gut microbiota for many years, recent advancements in molecular biology including next-generation sequencing technology has enabled researchers to gain new insight in this research field. While we are still far away from clearly understanding the exact roles and effecting modes of gut microbiota, growing evidence suggests that gut microbiota is important in modulating gut permeability and intestinal barrier functions. In this review, we summarize recent advances in the understanding of the leaky gut, bacterial translocation, and gut microbiota dysbiosis, with a particular focus on their association with extraintestinal autoimmune diseases, such as T1D and SLE.
The Intestinal Barrier
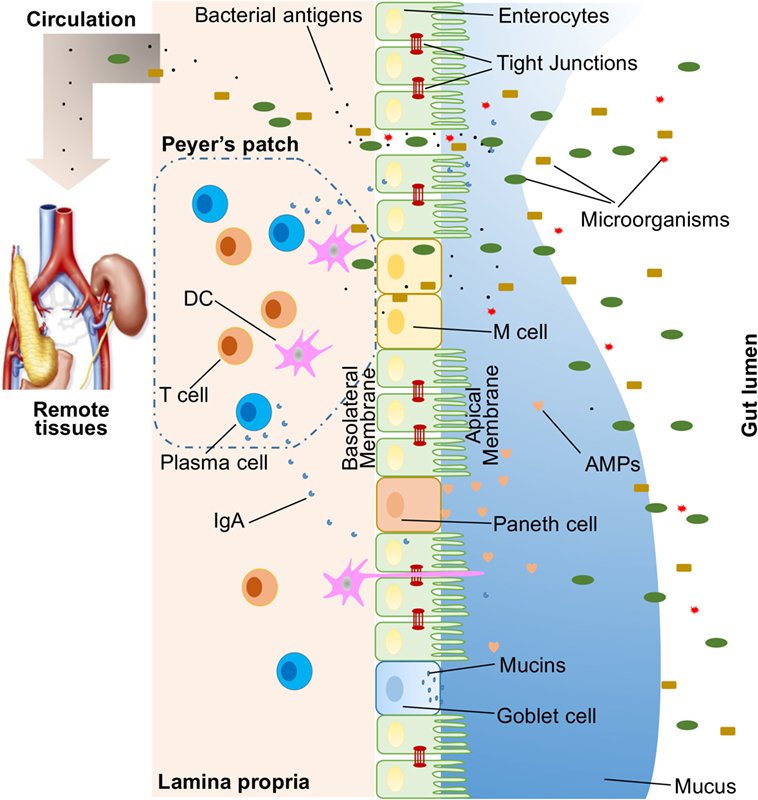
A large variety of exogenous substances colonize the gut lumen, such as microorganisms, toxins, and antigens. Without an intact and properly functioning intestinal barrier, these substances can penetrate the tissues beneath the intestinal epithelial lining, diffuse into blood and lymphatic circulations, and disrupt tissue homeostasis. However, there is an efficient multifaceted intestinal barrier system with physical, biochemical, and immunological components that prevents the entry of most pathogens (Figure 1). These components coordinate with each other to prevent uncontrolled translocation of luminal contents into the body. Below is a brief synopsis of the main components comprising the intestinal barrier.
FIGURE 1

Figure 1. Illustration of host intestinal barriers, including physical barrier (epithelium, tight junctions, mucus, commensal bacteria), biomedical barrier [antimicrobial proteins (AMPs)], and immunological barrier (lymphocytes and IgA). Also shown is the microbial translocation to remote tissues (for example, kidney and pancreas) in the presence of a leaky gut.
Physical Barrier
In humans, the intestinal epithelium covers as large as 400 m2 of surface area (1). Though only a single layer of cells, the intestinal epithelial cells (IECs) are the mainstay of the intestinal barrier and serve as a physical barrier (Figure 1). There are at least seven types of functional IECs—enterocytes, goblet cells, Paneth cells, microfold cells (M cells), enteroendocrine cells, cup cells, and tuft cells, although the functions of the last two cell populations are not well understood (10). Among all these cell types, enterocytes represent the absolute majority, accounting for at least 90% of crypt cells or villus cells. Enterocytes are absorptive cells and vital for nutrient uptake. However, growing evidence indicates that the functions of enterocytes are not limited to nutrient absorption. For example, enterocytes can control the abundance of Gram-positive bacteria by expressing RegIIIγ, one type of antimicrobial proteins (AMPs) (11–13). All epithelial cell types originate from Lgr5+ intestinal epithelial stem cells, which reside within the crypts (14). The turnover rate of IECs is high and the cells are renewed every 3–5 days in the mammalian intestine (10, 15), with the exception being the Paneth cells, which have a life span of about 2 months.
The IEC lining is continuous, and the contact between IECs is sealed by TJs (16). The paracellular pathway, in contrast to transcellular pathway, allows the transport of substances across the gut epithelium through the spaces between IECs. A large variety of molecules, mainly proteins, control the plasticity of TJs. More than 40 TJ proteins have been recognized, including occludin, claudins, junctional adhesion molecule A, and tricellulin (17). Under various pathological conditions, paracellular permeability may be increased, resulting in the entry of unwelcome, potentially harmful molecules.
On top of the gut epithelium, there are two layers of mucus, the inner and outer layers, that cover the whole intestinal epithelial lining and provide physical protection to separate luminal microorganisms from the epithelium. Organized by its major component, a highly glycosylated gel-forming mucin MUC2, the mucus contains diverse molecules including IgA as well as enzymes and proteins, such as lactoferrin (18). Goblet cells are the central cell type for the formation of mucus. They not only produce MUC2 mucin but also secret other mucus components such as ZG16, AGR2, FCGBP, CLCA1, and TFF3 (19, 20). Colitis would spontaneously develop in Muc2-deficient mice, indicating a critical role for MUC2 in mucosal protection (21). In addition to gel-forming mucins, there is another type of mucin that is in close proximity to epithelial cells, called transmembrane mucins. Enterocytes are the main producers of transmembrane mucins (20).
The gut commensal bacteria have been described as one component of the intestinal physical barrier primarily due to its two major functions (22). The first is to promote resistance to the colonization of harmful or pathogenic bacteria species by competing for nutrients, occupying attachment sites, and releasing antimicrobial substances (23, 24). Additionally, the gut microbiota regulates the digestion and absorption of nutrients to supply energy to epithelial cells, which are a major component of the physical barrier (25). A good example of the direct energy supply is the production of short-chain fatty acids by the gut microbiota, which are used by colonocytes for their development and metabolism (26). Taken together, IECs, the mucus layers, and gut microbial residents serve as the physical barrier to limit the entry of unfriendly luminal contents into host tissues.
Biochemical Barrier
Biochemical molecules with antimicrobial properties exist in the mucus as well as far into the lumen and include bile acids and AMPs (27, 28) (Figure 1). These diverse molecules form a complicated network to reduce the load of colonized bacteria and decrease the chance of contact between luminal antigens and host cells. They are a good supplement to the physical barrier and an essential component of the intestinal barrier function.
The proximal small intestine harbors very few microorganisms (29). But as the distance from the stomach increases, the pH rises and the number of colonized bacteria esculates (30). Facing a large number of microorganisms, which likely outnumber the number of host cells, multiple AMPs are generated to fight against invaders. These AMPs are divided into several types, including α- and β-defensins, C-type lectin, cathelicidin, lysozyme, and intestinal alkaline phosphatase (IAP) (27). Their detailed antimicrobial mechanisms are discussed elsewhere (31). As a major, but not exclusive, producer of AMPs, Paneth cells support and mediate the biochemical barrier function.
Immunological Barrier
Below the intestinal epithelium, there are organized lymphoid follicles, including the Peyer's patches and isolated lymphoid follicles. Inside the follicles, a variety of immune cells, including B cells, T cells, dendritic cells (DCs), and neutrophils, orchestrate the immune response by presenting antigens, secreting cytokines, and producing antigen-binding antibodies (Figure 1). In the intestinal epithelium where lymphoid follicles are found, M cells are present that transcytose antigens across the intestinal epithelium to the Peyer's patches underneath (14). In addition, goblet cells present acquired luminal antigens to CD103+ DCs in lamina propria in small intestine by forming goblet cell-associated antigen passages (GAPs) (32, 33). Interestingly, spontaneous antigen presentation was also observed in the colon, but only when the mice were raised germ-free (GF), or housed conventionally but with oral antibiotic treatment (34). This suggests that the antigen uptake process and formation of GAPs are regulated by the colonic microbiota (35). In addition, goblet cells and GAPs are capable of sensing invasive pathogens and inhibiting the translocation of pathogenic bacteria into the host immune system (36). Furthermore, intestinal mononuclear phagocytescan sense and sample luminal contents (37, 38). CX3CR1-expressing cells are responsible for this process, and antigen sampling is dependent on structures called transepithelial dendrites (TEDs) (39, 40). The formation of TEDs is regulated by CX3CR1+ macrophages and the expression of CX3CL1 by certain IECs (41, 42).
Another component of the immunological barrier is secretory IgA (SIgA). As the most abundant immunoglobulin in the body, IgA resides primarily on intestinal mucosal surfaces. While some people with selective IgA deficiency appear to be healthy, SIgA is important as it presumably interacts with commensal bacteria to provide protection against pathogens. A unique feature about SIgA is that is structurally resilient in protease-rich environments allowing it to remain functionally active compared to other antibody isotypes on mucosal surfaces (43). In adult humans, about 50 mg/kg of SIgA is produced daily by plasma cells residing in the intestinal lamina propria. Finally, SIgA can be transcytosed through the epithelium and secreted into the gut lumen.
Though not mentioned here, self-modulating factors, such as nerves and diverse cytokines, are also important for maintaining the normal functions of the intestinal barrier.
Gut Microbiota and the Intestinal Barrier
Microbiota can be sensed by the host through pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs). In the gut, the bacteria–host communications are largely dependent on the recognition of microbe-associated molecular patterns by PRRs expressed on immune and non-immune cells. Certain microbiota, bacterial products, and metabolites affect the intestinal barrier function and are responsible for the subsequent breakdown of tissue homeostasis. When there is a leaky gut, commensal bacteria in gut lumen, together with their products, are able to escape the lumen of the gut, which may induce inflammation and cause systemic tissue damages if translocated into peripheral circulation (Figure 1). This process of translocation is called microbial translocation (44).
Evidence from GF animals suggests that the development and function of the intestinal barrier are dependent on microbiota. In GF animals, due to the lack of bacterial stimulations, the thickness of the mucus layers is extremely reduced (45–48). The important role of gut microbiota in modulating mucin production from goblet cells is further evidenced in animals with lower loads of bacteria (49, 50). The thinner mucus layers would allow for bacteria penetration, which may initiate inflammation and inflammatory diseases such as colitis (46, 51). Commensal bacteria, or bacterial products such as lipopolysaccharide (LPS) and peptidoglycan, can restore the mucus layers (46, 47). A balance exists between commensal bacteria and the mucus layers, and together they contribute to the maintenance of gut homeostasis (48). Within the mucus layers, there are diverse secreted AMPs that can clear pathogens and control the colonization of commensal bacteria. Reciprocally, the production of some AMPs is regulated by microbiota and/or their products. For instance, RegIIIγ is the AMP necessary for physically separating commensal bacteria from intestinal epithelium (11). RegIIIγ has been shown to be suppressed in alcoholic patients and mice receiving ethanol treatment (52, 53). Prebiotics administration, or increasing probiotic Lactobacilli and Bifidobacteria, has been shown to restore the properties of RegIIIγ and control bacterial overgrowth (53). Ang4, a member of angiogenin family, is another example where gut commensals are known to modulate AMP production. In one study, Gordon and coworkers found that the production and secretion of Ang4 from mouse Paneth cells were induced by a predominant gut microflora, Bacteroides thetaiotaomicron (54). Therefore, the antibacterial activity of Ang4 against microbes in gut lumen is, in turn, dependent on the existence of certain commensal species.
In addition, an interaction exists between gut microbes and AMPs, such as IAP. Predominately produced by IECs, IAP is active either anchored on the epithelium membrane or secreted into gut lumen (55, 56). In IAP-deficient mice, it was noted that there were fewer microbes and an altered bacteria composition compared to control wild-type animals. In particular, the researchers noted a decrease in Lactobacillaceae (57, 58). Upregulated IAP activity can selectively increase LPS-suppressing bacteria (e.g., Bifidobacterium), while reducing LPS-producing bacteria (e.g., Escherichia coli) (59). Having the capacity to inactivate LPS in vivo, IAP is vital in preventing the translocation of LPS, the pro-inflammatory stimulus originated from bacteria (60, 61). Of note, the expression of IAP relies on the presence of microbiota. In GF zebrafish, the colonization of commensals, or even supplying LPS alone, could sufficiently induce IAP expression (62). It is worth mentioning that IAP can also regulate TJ proteins to enhance barrier function through increasing ZO-1, ZO-2, and occludin expression (63). Several others have also reported on the various types of AMPs and their function in the microbiota (64, 65).
Intestinal epithelial cells compose the single layer of intestinal epithelium, and the generation of new IECs from local intestinal stem cells is vital in maintaining the barrier function due to the high frequency of apoptosis and shedding of IECs (66). As much as 10% of all the gene transcriptions, especially genes related to immunity, cell proliferation, and metabolism, in IECs are regulated by gut microbiota (67). In GF and antibiotic-treated mice, epithelial proliferation rate is reduced, suggesting the role of microbiota on epithelium cell renewal (68, 69). LPS from E. coli can induce cell shedding in a dose-dependent manner (70, 71). Colonization of Bifidobacterium breve, or more precisely its surface component, exopolysaccharide, can positively modulate LPS-induced epithelium cell shedding through epithelial MyD88 signaling (70). The renewal of IECs relies on the activity of intestinal stem cells that are located at the base of crypts and express TLR4, the LPS receptor. TLR4 activation has been demonstrated to inhibit proliferation and promote the apoptosis of Lgr5+ intestinal stem cells. In mice bearing selective TLR4 deletion in intestinal stem cells, LPS is no longer able to inhibit the renewal of IECs (72). This process was found to be mediated by the p53-upregulated modulator of apoptosis (PUMA) as TLR4 activation in mice lacking PUMA was unaltered. Apart from LPS, bacterial metabolites, particularly butyrate, have also been identified as inhibitors of intestinal stem cell proliferation (73). The intestinal crypt architecture protects the intestinal stem cells from the negative effect of butyrate. As gatekeepers for the paracellular pathway, TJ complexes are also major targets of microbiota regulation (74). This is particularly true for certain probiotic species including, but not limited to, Lactobacillus rhamnosus (75–78), Streptococcus thermophilus (79), Lactobacillus reuteri (80), and Bifidobacterium infantis (81).
Mechanisms of Leaky Gut
A large variety of gut barrier disruptors and/or gut microbiota disturbers may potentially result in microbial translocation and subsequent inflammation locally and systemically. These include diet, infections, alcohol consumption, and burn injury.
Diet-Induced Gut Leakiness
Nutrients and food ingredients have been reported to contribute to the maintenance or alterations of gut microbiota and the intestinal barrier function (82). A recent review by De Santis et al. detailed many dietary factors that may modulate the intestinal barrier (83). Here, we review some recent publications and emphasize the effects of diet-induced alterations of gut microbiota on compromising the gut barrier function. Vitamin D (VD) has been recognized as an intestinal permeability protector by inducing the expression of TJ proteins ZO-1 and claudin-1. In VD receptor (VDR)-knockout mice, more severe experimental colitis has been observed, suggesting the protective effect of VD on the mucosal barrier (84). However, another group have recently found that VDR deficiency lowers, whereas VD treatment upregulates, the expression of claudin-2, a pore-forming TJ protein, which renders the intestinal epithelium leaky (85). Further analysis confirmed that VDR enhanced claudin-2 promoter activity. The exact role of VD and VDR on modulating intestinal permeability is therefore unclear and should be investigated carefully in association with gut microbiota. In a recent study by Desai et al., a low-fiber diet consumption was found to trigger the expansion of mucus-degrading bacteria, including Akkermansia muciniphila and Bacteroides caccae (45). As a result, the thickness of mucus is significantly decreased in mice fed with fiber-deficient diets, although the transcription of Muc2 gene was surprisingly heightened, possibly as a compensatory response. The thinner mucus and compromised intestinal barrier function lead to a higher susceptibility to certain colitis-causing pathogens (45). Moreover, a diet high in saturated fat has been shown to greatly decrease Lactobacillus and increase Oscillibacter, and these changes were correlated with significantly increased permeability in the proximal colon (86). Furthermore, studies revealed that the abundance of the Oscillospira genus was negatively correlated with the mRNA expression of barrier-forming TJ protein ZO-1.
Stress-Induced Gut Leakiness
Under certain circumstances, stress-induced alterations of gut microbiota and the impaired intestinal barrier would allow the occurrence of microbial translocation. Burn injury and alcohol consumption are examples of such stress. Burn injury results in increased intestinal permeability, which is mediated by increased activity of myosin light-chain (MLC) kinase (87, 88). It is known that MLC phosphorylation or kinase activation can trigger epithelial TJ opening (89–91). In burn injury, TJ proteins, including ZO-1, occluding, and claudin-1, are redistributed, which can be reversed by adding an MLC phosphorylation inhibitor (87). In addition, both humans and mice experiencing burn injury undergo similar alterations of gut microbiota, in particular, with increases of the abundance of bacteria from the Enterobacteriaceae family (88). Importantly, microbial translocation of these Gram-negative aerobic bacteria has been observed. Another research group, using a different burn injury mouse model reported increased colonic permeability together with reduced aerobic and anaerobic bacterial populations in the gut microbiota, particularly those producing butyrate (92). As a consequence, the butyrate level in the stool was significantly decreased in mice with burn injury. Interestingly, when the experimental mice received fecal microbiota transplant, their altered bacterial counts and impaired mucosal barrier function were reversed, suggesting direct involvement of microbiota in causing gut leakiness after burn injury.
Chronic alcohol consumption is responsible for intestinal barrier dysfunction, alterations on both the quality and quantity of gut microbiota, LPS translocation, and alcoholic liver disease (ALD). In both human and mouse, it has been well established that alcohol can disrupt intestinal barrier function, which is closely related to increased tumor necrosis factor (TNF) production from intestinal monocytes/macrophages and enterocytes bearing TNF-receptor 1, followed by downstream activation of MLC kinase (93). Notably, when mice given chronic alcohol also received oral antibiotic treatment, to remove the microbiota, the level of TNF production and intestinal permeability decreased to levels comparable to those in control mice (93). This indicates that the alcohol-induced, TNF-mediated gut leakiness is greatly dependent on gut microbiota. Indeed, though the mechanism is unknown, alcohol administration alters microbiota qualitatively and quantitatively in both human and mouse (94). Bacterial overgrowth has been observed with alcohol consumption, whereas antibiotics can decrease the bacterial load and attenuate ALD (53, 93, 95–97). Interestingly, probiotic Lactobacillus is significantly suppressed during alcohol consumption (53, 97). Directly supplying Lactobacillus strains or indirect stimulation of Lactobacilli with prebiotics or diets can decrease bacterial overgrowth, restore mucosal integrity of the intestine, and suppress microbial translocation (53, 94, 98, 99). Microbial translocation, especially the translocation of LPS, is involved in ALD development and progression as evidenced by the lack of ALD in mice deficient of TLR4 (100, 101). It is worth noting that some bacteria species can produce alcohol, including E. coli and Weissella confusa, and this may be the mechanism by which they compromise the intestinal barrier function (102, 103).
Infections can play a role in regulating the mucosal barrier. A good example is Helicobacter pylori, a Gram-negative bacterium infecting the human stomach (104). H. pylori is known to directly increase epithelial permeability by redistributing TJ protein ZO-1 (105, 106). In addition, bacteriophages, which are usually not considered pathogenic to mammals, can have an impact on the leaky gut. When rats were given a bacteriophage cocktail containing phages against Salmonella enterica, disruption of the intestinal barrier integrity was observed (107). The authors speculated that the gut microbiota might have been affected by bacteriophages, but sequencing data were not supplied to support their claims.
Taken together, perturbation of gut microbiota, which may be the consequence of diverse interventions, can lead to increased intestinal permeability and translocation of bacterial components and products. Such microbial translocation can subsequently trigger an abnormal immune response, causing inflammation and/or tissue damage in extraintestinal organs.
Leaky Gut and Autoimmune Disorders
Several disease states have been associated with gut microbiota dysbiosis, intestinal barrier dysfunction, and microbial translocation. These include Alzheimer's disease, ALD, cancer, and multiple autoimmune disorders. Autoimmune disorders are characterized by the generation of autoantibodies against self-antigens that attack the body's own tissues, resulting in damage. Genetic and environmental triggers have been long known as the major contributors to the development of autoimmunity. Increasing evidence in recent years suggests that microbial translocation and intestinal barrier dysfunction, which may be affected by gut microbiota, are another important causative element for autoimmune disorders (2–6). T1D and SLE are examples discussed below that reveal advancements in the understanding of the mechanisms behind the interaction between the leaky gut and autoimmune disorders.
Type 1 Diabetes
Type 1 diabetes is an organ-specific autoimmune disorder characterized by an autoimmune response against the host's own pancreatic β cells, leading to insufficient insulin production from the pancreas (108). Some argue that the leaky gut is only an outcome of disease progression rather than an initiator or exacerbator of disease (109), but this should not be the case for T1D. This is supported by the following evidences. First, studies utilizing human subjects affected by T1D or T1D-prone animal models have indicated that impaired intestinal barrier function occurs before disease onset (110–112). Second, the pathogenic role that increased intestinal permeability plays in T1D is zonulin-dependent, and the production of zonulin relies on bacterial colonization (113). Reversion of intestinal barrier dysbiosis by adding a zonulin inhibitor ameliorated T1D manifestations in disease-prone rats (114). Third, a recent study has provided evidence that microbial translocation contributes to T1D development (115). In streptozotocin-induced T1D, mice treated with streptozotocin harbor a distinct microbiota compared to vehicle-treated controls. Importantly, gut bacteria were shown to be able to translocate into pancreatic lymph nodes (PLNs) and contribute to T1D development (115). When mice were treated with oral antibiotics, PLNs appeared to be sterile and the disease was attenuated. Further analysis revealed that the translocated bacteria in PLNs triggered NOD2 activation and exacerbated T1D. Altogether, these results suggest an essential role for the leaky gut in driving the progression of T1D.
Systemic Lupus Erythematosus
Systemic lupus erythematosus, or lupus, is an autoimmune disorder characterized by severe and persistent inflammation that leads to tissue damage in multiple organs (116). Although SLE affects both men and women, women of childbearing age are diagnosed about nine times more often than men. LPS, a cell wall component of Gram-negative bacteria, can promote SLE development and disease progression upon penetration of the intestinal epithelium and translocation into tissues (117). In SLE patients, the higher level of soluble CD14 suggests an increase in LPS, as soluble CD14 is released from monocytes when the cells are exposed to LPS (118). Activation of TLR4 exacerbates lupus development (119–121). Mice spontaneously develop lupus when TLR4 responsiveness is increased, whereas the exacerbated disease phenotype can be significantly ameliorated when the commensal gut flora is removed by antibiotic treatment (121). This clearly indicates that TLR4 hyperresponsiveness to gut flora (which contains LPS) contributes to the pathogenesis of SLE. Moreover, the development of lupus in wild-type mice (C57BL/6 or BALB/c) immunized with phospholipid-binding proteins can be facilitated by the administration of LPS (122–124). Conversely, inhibition of TLR4 results in reduced autoantibody production and lowered renal glomerular IgG deposits in lupus-prone mice (125, 126). Taken together, these data suggest that LPS stimulation and TLR4 activation as disease-initiating factors for SLE. Lipoteichoic acid (LTA), a component of the Gram-positive bacterial cell wall, can also promote lupus disease. The expression of TLR2, the receptor of LTA, has been reported to be increased in SLE patients (127). In lupus-prone mice, TLR2 activation triggers lupus nephritis, whereas TLR2 knockout attenuates lupus-like symptoms (125, 128–130). Recently, another bacterial antigen that may mimic self-antigens has been recognized to induce autoantibody production (131).
Several downstream proteins in the TLR signaling cascade are highly relevant to the pathogenesis of SLE and are potential therapeutic targets, including MyD88, IRAKs, and IFNα (132). Deficiency of MyD88, in particular, has been shown to ameliorate lupus disease in MRL/lpr mice (133, 134), suggesting a potential role for TLRs to communicate with harmful bacteria in the gut microbiota. Conversely, there is a paucity of data pertaining to members of the NLR family. The most extensively characterized NLRs are associated with inflammasome formation (135, 136). Loss of NLRP3 and AIM2 inflammasome function was found to significantly contribute to lupus pathogenesis (137). Interestingly, both of these inflammasomes were found compromised in NZB mice, a lupus-prone model. Consistent with this finding, loss of ASC (apoptosis-associated speck-like protein containing CARD), a common adaptor protein required for inflammasome formation in B6-Faslpr mice led to exacerbation of lupus-like disease (138). These results suggest a potential role for NLRs to recognize protective bacteria in the gut microbiota. Therefore, it appears that TLRs and NLRs make distinct contributions to lupus pathogenesis by sensing harmful and protective bacteria, respectively. Both types of bacteria can come from gut microbiota through microbial translocation, especially in the presence of a leaky gut.
Reversing the Leaky Gut as a Potential Therapy
Considering the contributions of leaky gut and bacterial translocation to inflammation and multiple diseases, reversing gut leakiness appears to be an attractive therapeutic strategy. Prebiotics and probiotics, for example, can be used to reduce intestinal permeability (139). Diverse probiotic species have been uncovered that possess the properties to protect the intestinal barrier through targeting different components of the mucosal barrier system. The human commensal Bacteroides fragilis may serve as such a probiotic (140). In a mouse model, autism spectrum disorder (ASD) has been shown to be accompanied by intestinal barrier dysfunction, gut microbiota dysbiosis, and leakiness of 4-ethylphenylsulfate (4EPS), which originates from the commensal bacteria. When 4EPS was given to wild-type mice, it directly caused behavioral abnormalities similar to ASD mice. Treatment with B. fragilis reduced the translocation of disease-causative 4EPS, and significantly ameliorated the behavior defects. The therapeutic benefit of B. fragilis is believed to be due to its ability to alter microbial composition and enhance intestinal barrier function (140). B. fragilis is also known for its capability to induce the development of Foxp3+ regulatory T cells, a process regulated by another product of B. fragilis, polysaccharide A (PSA) (141, 142). B. fragilis and PSA are beneficial against inflammatory diseases, such as colitis and experimental autoimmune encephalomyelitis (141, 143). The application of B. fragilis to prevent the leaky gut and reverse autoimmunity warrants further investigation. In a practical point of view, probiotic candidates with different targets on reversing the leaky gut may synergistically act to attenuate disease as thus may serve as a probiotic cocktail. As probiotics are generally considered safe, it is anticipated that they will become cost-effective treatment options for people with autoimmune diseases in the foreseeable future. This is a very young but exciting field in which much still remains to be learned.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Preparation of this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R03AI117597. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
2. Lin R, Zhou L, Zhang J, Wang B. Abnormal intestinal permeability and microbiota in patients with autoimmune hepatitis. Int J Clin Exp Pathol (2015) 8(5):5153–60.
PubMed Abstract | Google Scholar
3. Khaleghi S, Ju JM, Lamba A, Murray JA. The potential utility of tight junction regulation in celiac disease: focus on larazotide acetate. Therap Adv Gastroenterol (2016) 9(1):37–49. doi:10.1177/1756283X15616576
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Fasano A, Shea-Donohue T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Pract Gastroenterol Hepatol (2005) 2(9):416–22. doi:10.1038/ncpgasthep0259
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Tlaskalova-Hogenova H, Stepankova R, Kozakova H, Hudcovic T, Vannucci L, Tuckova L, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol (2011) 8(2):110–20. doi:10.1038/cmi.2010.67
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science (2011) 334(6053):255–8. doi:10.1126/science.1209791
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A (2008) 105(52):20858–63. doi:10.1073/pnas.0808723105
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Mabbott NA, Donaldson DS, Ohno H, Williams IR, Mahajan A. Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol (2013) 6(4):666–77. doi:10.1038/mi.2013.30
PubMed Abstract | CrossRef Full Text | Google Scholar
15. van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol (2009) 71:241–60. doi:10.1146/annurev.physiol.010908.163145
CrossRef Full Text | Google Scholar
16. Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol (2009) 1(2):a002584. doi:10.1101/cshperspect.a002584
CrossRef Full Text | Google Scholar
17. Yamazaki Y, Okawa K, Yano T, Tsukita S, Tsukita S. Optimized proteomic analysis on gels of cell-cell adhering junctional membrane proteins. Biochemistry (2008) 47(19):5378–86. doi:10.1021/bi8002567
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Pelaseyed T, Bergstrom JH, Gustafsson JK, Ermund A, Birchenough GM, Schutte A, et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol Rev (2014) 260(1):8–20. doi:10.1111/imr.12182
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology (2006) 131(1):117–29. doi:10.1053/j.gastro.2006.04.020
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev (2010) 90(3):859–904. doi:10.1152/physrev.00045.2009
CrossRef Full Text | Google Scholar
24. Ming L, Zhang Q, Yang L, Huang JA. Comparison of antibacterial effects between antimicrobial peptide and bacteriocins isolated from Lactobacillus plantarum on three common pathogenic bacteria. Int J Clin Exp Med (2015) 8(4):5806–11.
PubMed Abstract | Google Scholar
27. Dupont A, Heinbockel L, Brandenburg K, Hornef MW. Antimicrobial peptides and the enteric mucus layer act in concert to protect the intestinal mucosa. Gut Microbes (2014) 5(6):761–5. doi:10.4161/19490976.2014.972238
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Hofmann AF, Eckmann L. How bile acids confer gut mucosal protection against bacteria. Proc Natl Acad Sci U S A (2006) 103(12):4333–4. doi:10.1073/pnas.0600780103
CrossRef Full Text | Google Scholar
32. Howe SE, Lickteig DJ, Plunkett KN, Ryerse JS, Konjufca V. The uptake of soluble and particulate antigens by epithelial cells in the mouse small intestine. PLoS One (2014) 9(1):e86656. doi:10.1371/journal.pone.0086656
PubMed Abstract | CrossRef Full Text | Google Scholar
33. McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature (2012) 483(7389):345–9. doi:10.1038/nature10863
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Knoop KA, McDonald KG, McCrate S, McDole JR, Newberry RD. Microbial sensing by goblet cells controls immune surveillance of luminal antigens in the colon. Mucosal Immunol (2015) 8(1):198–210. doi:10.1038/mi.2014.58
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Birchenough GM, Johansson ME, Gustafsson JK, Bergstrom JH, Hansson GC. New developments in goblet cell mucus secretion and function. Mucosal Immunol (2015) 8(4):712–9. doi:10.1038/mi.2015.32
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Knoop KA, McDonald KG, Kulkarni DH, Newberry RD. Antibiotics promote inflammation through the translocation of native commensal colonic bacteria. Gut (2016) 65(7):1100–9. doi:10.1136/gutjnl-2014-309059
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol (2001) 2(4):361–7. doi:10.1038/86373
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Vallon-Eberhard A, Landsman L, Yogev N, Verrier B, Jung S. Transepithelial pathogen uptake into the small intestinal lamina propria. J Immunol (2006) 176(4):2465–9. doi:10.4049/jimmunol.176.4.2465
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, et al. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature (2013) 494(7435):116–20. doi:10.1038/nature11809
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science (2005) 307(5707):254–8. doi:10.1126/science.1102901
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Kim KW, Vallon-Eberhard A, Zigmond E, Farache J, Shezen E, Shakhar G, et al. In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood (2011) 118(22):e156–67. doi:10.1182/blood-2011-04-348946
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell (2016) 167(5):1339–53 e21. doi:10.1016/j.cell.2016.10.043
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Johansson ME, Jakobsson HE, Holmen-Larsson J, Schutte A, Ermund A, Rodriguez-Pineiro AM, et al. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe (2015) 18(5):582–92. doi:10.1016/j.chom.2015.10.007
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Petersson J, Schreiber O, Hansson GC, Gendler SJ, Velcich A, Lundberg JO, et al. Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am J Physiol Gastrointest Liver Physiol (2011) 300(2):G327–33. doi:10.1152/ajpgi.00422.2010
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Wrzosek L, Miquel S, Noordine ML, Bouet S, Joncquel Chevalier-Curt M, Robert V, et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol (2013) 11:61. doi:10.1186/1741-7007-11-61
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Forder RE, Howarth GS, Tivey DR, Hughes RJ. Bacterial modulation of small intestinal goblet cells and mucin composition during early posthatch development of poultry. Poult Sci (2007) 86(11):2396–403. doi:10.3382/ps.2007-00222
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Wlodarska M, Willing B, Keeney KM, Menendez A, Bergstrom KS, Gill N, et al. Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infect Immun (2011) 79(4):1536–45. doi:10.1128/IAI.01104-10
PubMed Abstract | CrossRef Full Text | Google Scholar
51. Johansson ME, Gustafsson JK, Sjoberg KE, Petersson J, Holm L, Sjovall H, et al. Bacteria penetrate the inner mucus layer before inflammation in the dextran sulfate colitis model. PLoS One (2010) 5(8):e12238. doi:10.1371/journal.pone.0012238
CrossRef Full Text | Google Scholar
52. Wang L, Fouts DE, Starkel P, Hartmann P, Chen P, Llorente C, et al. Intestinal REG3 lectins protect against alcoholic steatohepatitis by reducing mucosa-associated microbiota and preventing bacterial translocation. Cell Host Microbe (2016) 19(2):227–39. doi:10.1016/j.chom.2016.01.003
PubMed Abstract | CrossRef Full Text | Google Scholar
53. Yan AW, Fouts DE, Brandl J, Starkel P, Torralba M, Schott E, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology (2011) 53(1):96–105. doi:10.1002/hep.24018
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Eliakim R, Mahmood A, Alpers DH. Rat intestinal alkaline phosphatase secretion into lumen and serum is coordinately regulated. Biochim Biophys Acta (1991) 1091(1):1–8. doi:10.1016/0167-4889(91)90213-H
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Nakano T, Inoue I, Alpers DH, Akiba Y, Katayama S, Shinozaki R, et al. Role of lysophosphatidylcholine in brush-border intestinal alkaline phosphatase release and restoration. Am J Physiol Gastrointest Liver Physiol (2009) 297(1):G207–14. doi:10.1152/ajpgi.90590.2008
PubMed Abstract | CrossRef Full Text | Google Scholar
57. Malo MS, Alam SN, Mostafa G, Zeller SJ, Johnson PV, Mohammad N, et al. Intestinal alkaline phosphatase preserves the normal homeostasis of gut microbiota. Gut (2010) 59(11):1476–84. doi:10.1136/gut.2010.211706
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut (2006) 55(2):205–11. doi:10.1136/gut.2005.073817
PubMed Abstract | CrossRef Full Text | Google Scholar
59. Kaliannan K, Wang B, Li XY, Kim KJ, Kang JX. A host-microbiome interaction mediates the opposing effects of omega-6 and omega-3 fatty acids on metabolic endotoxemia. Sci Rep (2015) 5:11276. doi:10.1038/srep11276
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe (2007) 2(6):371–82. doi:10.1016/j.chom.2007.10.010
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Yang Y, Millan JL, Mecsas J, Guillemin K. Intestinal alkaline phosphatase deficiency leads to lipopolysaccharide desensitization and faster weight gain. Infect Immun (2015) 83(1):247–58. doi:10.1128/IAI.02520-14
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Bates JM, Mittge E, Kuhlman J, Baden KN, Cheesman SE, Guillemin K. Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev Biol (2006) 297(2):374–86. doi:10.1016/j.ydbio.2006.05.006
PubMed Abstract | CrossRef Full Text | Google Scholar
63. Liu W, Hu D, Huo H, Zhang W, Adiliaghdam F, Morrison S, et al. Intestinal alkaline phosphatase regulates tight junction protein levels. J Am Coll Surg (2016) 222(6):1009–17. doi:10.1016/j.jamcollsurg.2015.12.006
PubMed Abstract | CrossRef Full Text | Google Scholar
66. Williams JM, Duckworth CA, Burkitt MD, Watson AJ, Campbell BJ, Pritchard DM. Epithelial cell shedding and barrier function: a matter of life and death at the small intestinal villus tip. Vet Pathol (2015) 52(3):445–55. doi:10.1177/0300985814559404
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Sommer F, Nookaew I, Sommer N, Fogelstrand P, Backhed F. Site-specific programming of the host epithelial transcriptome by the gut microbiota. Genome Biol (2015) 16:62. doi:10.1186/s13059-015-0614-4
PubMed Abstract | CrossRef Full Text | Google Scholar
68. Reikvam DH, Erofeev A, Sandvik A, Grcic V, Jahnsen FL, Gaustad P, et al. Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS One (2011) 6(3):e17996. doi:10.1371/journal.pone.0017996
PubMed Abstract | CrossRef Full Text | Google Scholar
69. Stedman A, Nigro G, Sansonetti PJ. [Microbiota-intestinal stem cells dialog: a key element for intestinal regeneration]. Med Sci (Paris) (2016) 32(11):983–90. doi:10.1051/medsci/20163211014
PubMed Abstract | CrossRef Full Text | Google Scholar
70. Hughes KR, Harnisch LC, Alcon-Giner C, Mitra S, Wright CJ, Ketskemety J, et al. Bifidobacterium breve reduces apoptotic epithelial cell shedding in an exopolysaccharide and MyD88-dependent manner. Open Biol (2017) 7(1):160155. doi:10.1098/rsob.160155
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Williams JM, Duckworth CA, Watson AJ, Frey MR, Miguel JC, Burkitt MD, et al. A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Dis Model Mech (2013) 6(6):1388–99. doi:10.1242/dmm.013284
PubMed Abstract | CrossRef Full Text | Google Scholar
72. Neal MD, Sodhi CP, Jia H, Dyer M, Egan CE, Yazji I, et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J Biol Chem (2012) 287(44):37296–308. doi:10.1074/jbc.M112.375881
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, et al. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell (2016) 167(4):1137. doi:10.1016/j.cell.2016.10.034
CrossRef Full Text | Google Scholar
74. Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr (2011) 141(5):769–76. doi:10.3945/jn.110.135657
PubMed Abstract | CrossRef Full Text | Google Scholar
75. Donato KA, Gareau MG, Wang YJ, Sherman PM. Lactobacillus rhamnosus GG attenuates interferon-{gamma} and tumour necrosis factor-alpha-induced barrier dysfunction and pro-inflammatory signalling. Microbiology (2010) 156(Pt 11):3288–97. doi:10.1099/mic.0.040139-0
PubMed Abstract | CrossRef Full Text | Google Scholar
76. Johnson-Henry KC, Donato KA, Shen-Tu G, Gordanpour M, Sherman PM. Lactobacillus rhamnosus strain GG prevents enterohemorrhagic Escherichia coli O157: H7-induced changes in epithelial barrier function. Infect Immun (2008) 76(4):1340–8. doi:10.1128/IAI.00778-07
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Miyauchi E, Morita H, Tanabe S. Lactobacillus rhamnosus alleviates intestinal barrier dysfunction in part by increasing expression of zonula occludens-1 and myosin light-chain kinase in vivo. J Dairy Sci (2009) 92(6):2400–8. doi:10.3168/jds.2008-1698
PubMed Abstract | CrossRef Full Text | Google Scholar
78. Seth A, Yan F, Polk DB, Rao RK. Probiotics ameliorate the hydrogen peroxide-induced epithelial barrier disruption by a PKC- and MAP kinase-dependent mechanism. Am J Physiol Gastrointest Liver Physiol (2008) 294(4):G1060–9. doi:10.1152/ajpgi.00202.2007
PubMed Abstract | CrossRef Full Text | Google Scholar
79. Resta-Lenert S, Barrett KE. Live probiotics protect intestinal epithelial cells from the effects of infection with enteroinvasive Escherichia coli (EIEC). Gut (2003) 52(7):988–97. doi:10.1136/gut.52.7.988
PubMed Abstract | CrossRef Full Text | Google Scholar
80. Ewaschuk JB, Diaz H, Meddings L, Diederichs B, Dmytrash A, Backer J, et al. Secreted bioactive factors from Bifidobacterium infantis enhance epithelial cell barrier function. Am J Physiol Gastrointest Liver Physiol (2008) 295(5):G1025–34. doi:10.1152/ajpgi.90227.2008
PubMed Abstract | CrossRef Full Text | Google Scholar
84. Kong J, Zhang Z, Musch MW, Ning G, Sun J, Hart J, et al. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am J Physiol Gastrointest Liver Physiol (2008) 294(1):G208–16. doi:10.1152/ajpgi.00398.2007
PubMed Abstract | CrossRef Full Text | Google Scholar
86. Lam YY, Ha CW, Campbell CR, Mitchell AJ, Dinudom A, Oscarsson J, et al. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLoS One (2012) 7(3):e34233. doi:10.1371/journal.pone.0034233
PubMed Abstract | CrossRef Full Text | Google Scholar
88. Earley ZM, Akhtar S, Green SJ, Naqib A, Khan O, Cannon AR, et al. Burn injury alters the intestinal microbiome and increases gut permeability and bacterial translocation. PLoS One (2015) 10(7):e0129996. doi:10.1371/journal.pone.0129996
PubMed Abstract | CrossRef Full Text | Google Scholar
92. Kuethe JW, Armocida SM, Midura EF, Rice TC, Hildeman DA, Healy DP, et al. Fecal microbiota transplant restores mucosal integrity in a murine model of burn injury. Shock (2016) 45(6):647–52. doi:10.1097/SHK.0000000000000551
PubMed Abstract | CrossRef Full Text | Google Scholar
93. Chen P, Starkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology (2015) 61(3):883–94. doi:10.1002/hep.27489
PubMed Abstract | CrossRef Full Text | Google Scholar
94. Chen P, Torralba M, Tan J, Embree M, Zengler K, Starkel P, et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology (2015) 148(1):203–14 e16. doi:10.1053/j.gastro.2014.09.014
PubMed Abstract | CrossRef Full Text | Google Scholar
95. Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology (1995) 108(1):218–24. doi:10.1016/0016-5085(95)90027-6
PubMed Abstract | CrossRef Full Text | Google Scholar
96. Casafont Morencos F, de las Heras Castano G, Martin Ramos L, Lopez Arias MJ, Ledesma F, Pons Romero F. Small bowel bacterial overgrowth in patients with alcoholic cirrhosis. Dig Dis Sci (1996) 41(3):552–6. doi:10.1007/BF02282340
PubMed Abstract | CrossRef Full Text | Google Scholar
97. Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA, et al. Colonic microbiome is altered in alcoholism. Am J Physiol Gastrointest Liver Physiol (2012) 302(9):G966–78. doi:10.1152/ajpgi.00380.2011
PubMed Abstract | CrossRef Full Text | Google Scholar
98. Forsyth CB, Farhadi A, Jakate SM, Tang Y, Shaikh M, Keshavarzian A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol (2009) 43(2):163–72. doi:10.1016/j.alcohol.2008.12.009
PubMed Abstract | CrossRef Full Text | Google Scholar
99. Wang Y, Kirpich I, Liu Y, Ma Z, Barve S, McClain CJ, et al. Lactobacillus rhamnosus GG treatment potentiates intestinal hypoxia-inducible factor, promotes intestinal integrity and ameliorates alcohol-induced liver injury. Am J Pathol (2011) 179(6):2866–75. doi:10.1016/j.ajpath.2011.08.039
PubMed Abstract | CrossRef Full Text | Google Scholar
100. Parlesak A, Schafer C, Schutz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol (2000) 32(5):742–7. doi:10.1016/S0168-8278(00)80242-1
PubMed Abstract | CrossRef Full Text | Google Scholar
101. Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology (2001) 34(1):101–8. doi:10.1053/jhep.2001.25350
PubMed Abstract | CrossRef Full Text | Google Scholar
102. Elshaghabee FM, Bockelmann W, Meske D, de Vrese M, Walte HG, Schrezenmeir J, et al. Ethanol production by selected intestinal microorganisms and lactic acid bacteria growing under different nutritional conditions. Front Microbiol (2016) 7:47. doi:10.3389/fmicb.2016.00047
PubMed Abstract | CrossRef Full Text | Google Scholar
103. Tyler AD, Knox N, Kabakchiev B, Milgrom R, Kirsch R, Cohen Z, et al. Characterization of the gut-associated microbiome in inflammatory pouch complications following ileal pouch-anal anastomosis. PLoS One (2013) 8(9):e66934. doi:10.1371/journal.pone.0066934
PubMed Abstract | CrossRef Full Text | Google Scholar
104. Yu QH, Yang Q. Diversity of tight junctions (TJs) between gastrointestinal epithelial cells and their function in maintaining the mucosal barrier. Cell Biol Int (2009) 33(1):78–82. doi:10.1016/j.cellbi.2008.09.007
PubMed Abstract | CrossRef Full Text | Google Scholar
105. Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science (2003) 300(5624):1430–4. doi:10.1126/science.1081919
PubMed Abstract | CrossRef Full Text | Google Scholar
106. Fedwick JP, Lapointe TK, Meddings JB, Sherman PM, Buret AG. Helicobacter pylori activates myosin light-chain kinase to disrupt claudin-4 and claudin-5 and increase epithelial permeability. Infect Immun (2005) 73(12):7844–52. doi:10.1128/IAI.73.12.7844-7852.2005
PubMed Abstract | CrossRef Full Text | Google Scholar
110. Damci T, Nuhoglu I, Devranoglu G, Osar Z, Demir M, Ilkova H. Increased intestinal permeability as a cause of fluctuating postprandial blood glucose levels in type 1 diabetic patients. Eur J Clin Invest (2003) 33(5):397–401. doi:10.1046/j.1365-2362.2003.01161.x
PubMed Abstract | CrossRef Full Text | Google Scholar
111. Meddings JB, Jarand J, Urbanski SJ, Hardin J, Gall DG. Increased gastrointestinal permeability is an early lesion in the spontaneously diabetic BB rat. Am J Physiol (1999) 276(4 Pt 1):G951–7.
PubMed Abstract | Google Scholar
112. Secondulfo M, Iafusco D, Carratu R, deMagistris L, Sapone A, Generoso M, et al. Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac, type I diabetic patients. Dig Liver Dis (2004) 36(1):35–45. doi:10.1016/j.dld.2003.09.016
PubMed Abstract | CrossRef Full Text | Google Scholar
113. El Asmar R, Panigrahi P, Bamford P, Berti I, Not T, Coppa GV, et al. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology (2002) 123(5):1607–15. doi:10.1053/gast.2002.36578
PubMed Abstract | CrossRef Full Text | Google Scholar
114. Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, et al. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci U S A (2005) 102(8):2916–21. doi:10.1073/pnas.0500178102
PubMed Abstract | CrossRef Full Text | Google Scholar
115. Costa FR, Francozo MC, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med (2016) 213(7):1223–39. doi:10.1084/jem.20150744
PubMed Abstract | CrossRef Full Text | Google Scholar
118. Nockher WA, Wigand R, Schoeppe W, Scherberich JE. Elevated levels of soluble CD14 in serum of patients with systemic lupus erythematosus. Clin Exp Immunol (1994) 96(1):15–9. doi:10.1111/j.1365-2249.1994.tb06222.x
PubMed Abstract | CrossRef Full Text | Google Scholar
119. Lee TP, Huang JC, Liu CJ, Chen HJ, Chen YH, Tsai YT, et al. Interactions of surface-expressed TLR-4 and endosomal TLR-9 accelerate lupus progression in anti-dsDNA antibody transgenic mice. Exp Biol Med (Maywood) (2014) 239(6):715–23. doi:10.1177/1535370214525299
PubMed Abstract | CrossRef Full Text | Google Scholar
120. Lee TP, Tang SJ, Wu MF, Song YC, Yu CL, Sun KH. Transgenic overexpression of anti-double-stranded DNA autoantibody and activation of Toll-like receptor 4 in mice induce severe systemic lupus erythematosus syndromes. J Autoimmun (2010) 35(4):358–67. doi:10.1016/j.jaut.2010.07.007
PubMed Abstract | CrossRef Full Text | Google Scholar
121. Liu B, Yang Y, Dai J, Medzhitov R, Freudenberg MA, Zhang PL, et al. TLR4 up-regulation at protein or gene level is pathogenic for lupus-like autoimmune disease. J Immunol (2006) 177(10):6880–8. doi:10.4049/jimmunol.177.10.6880
PubMed Abstract | CrossRef Full Text | Google Scholar
122. Levine JS, Subang R, Nasr SH, Fournier S, Lajoie G, Wither J, et al. Immunization with an apoptotic cell-binding protein recapitulates the nephritis and sequential autoantibody emergence of systemic lupus erythematosus. J Immunol (2006) 177(9):6504–16. doi:10.4049/jimmunol.177.9.6504
PubMed Abstract | CrossRef Full Text | Google Scholar
123. Levine JS, Subang R, Setty S, Cabrera J, Laplante P, Fritzler MJ, et al. Phospholipid-binding proteins differ in their capacity to induce autoantibodies and murine systemic lupus erythematosus. Lupus (2014) 23(8):752–68. doi:10.1177/0961203314525676
PubMed Abstract | CrossRef Full Text | Google Scholar
124. Tolomeo T, Rico De Souza A, Roter E, Dieude M, Amireault P, Subang R, et al. T cells demonstrate a Th1-biased response to native beta2-glycoprotein I in a murine model of anti-phospholipid antibody induction. Autoimmunity (2009) 42(4):292–5. doi:10.1080/08916930902828254
CrossRef Full Text | Google Scholar
125. Lartigue A, Colliou N, Calbo S, Francois A, Jacquot S, Arnoult C, et al. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J Immunol (2009) 183(10):6207–16. doi:10.4049/jimmunol.0803219
PubMed Abstract | CrossRef Full Text | Google Scholar
126. Summers SA, Hoi A, Steinmetz OM, O'Sullivan KM, Ooi JD, Odobasic D, et al. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun (2010) 35(4):291–8. doi:10.1016/j.jaut.2010.05.004
PubMed Abstract | CrossRef Full Text | Google Scholar
127. Liu Y, Liao J, Zhao M, Wu H, Yung S, Chan TM, et al. Increased expression of TLR2 in CD4(+) T cells from SLE patients enhances immune reactivity and promotes IL-17 expression through histone modifications. Eur J Immunol (2015) 45(9):2683–93. doi:10.1002/eji.201445219
PubMed Abstract | CrossRef Full Text | Google Scholar
128. Leiss H, Niederreiter B, Bandur T, Schwarzecker B, Bluml S, Steiner G, et al. Pristane-induced lupus as a model of human lupus arthritis: evolvement of autoantibodies, internal organ and joint inflammation. Lupus (2013) 22(8):778–92. doi:10.1177/0961203313492869
PubMed Abstract | CrossRef Full Text | Google Scholar
129. Pawar RD, Castrezana-Lopez L, Allam R, Kulkarni OP, Segerer S, Radomska E, et al. Bacterial lipopeptide triggers massive albuminuria in murine lupus nephritis by activating toll-like receptor 2 at the glomerular filtration barrier. Immunology (2009) 128(1 Suppl):e206–21. doi:10.1111/j.1365-2567.2008.02948.x
PubMed Abstract | CrossRef Full Text | Google Scholar
130. Urbonaviciute V, Starke C, Pirschel W, Pohle S, Frey S, Daniel C, et al. Toll-like receptor 2 is required for autoantibody production and development of renal disease in pristane-induced lupus. Arthritis Rheum (2013) 65(6):1612–23. doi:10.1002/art.37914
PubMed Abstract | CrossRef Full Text | Google Scholar
131. Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA composites of bacterial biofilms stimulate autoimmunity. Immunity (2015) 42(6):1171–84. doi:10.1016/j.immuni.2015.06.002
PubMed Abstract | CrossRef Full Text | Google Scholar
132. Wu YW, Tang W, Zuo JP. Toll-like receptors: potential targets for lupus treatment. Acta Pharmacol Sin (2015) 36(12):1395–407. doi:10.1038/aps.2015.91
CrossRef Full Text | Google Scholar
133. Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity (2013) 38(3):528–40. doi:10.1016/j.immuni.2012.11.017
PubMed Abstract | CrossRef Full Text | Google Scholar
134. Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol (2010) 184(4):1840–8. doi:10.4049/jimmunol.0902592
PubMed Abstract | CrossRef Full Text | Google Scholar
137. Sester DP, Sagulenko V, Thygesen SJ, Cridland JA, Loi YS, Cridland SO, et al. Deficient NLRP3 and AIM2 inflammasome function in autoimmune NZB mice. J Immunol (2015) 195(3):1233–41. doi:10.4049/jimmunol.1402859
PubMed Abstract | CrossRef Full Text | Google Scholar
138. Lech M, Lorenz G, Kulkarni OP, Grosser MO, Stigrot N, Darisipudi MN, et al. NLRP3 and ASC suppress lupus-like autoimmunity by driving the immunosuppressive effects of TGF-beta receptor signalling. Ann Rheum Dis (2015) 74(12):2224–35. doi:10.1136/annrheumdis-2014-205496
CrossRef Full Text | Google Scholar
139. Lamprecht M, Bogner S, Schippinger G, Steinbauer K, Fankhauser F, Hallstroem S, et al. Probiotic supplementation affects markers of intestinal barrier, oxidation, and inflammation in trained men; a randomized, double-blinded, placebo-controlled trial. J Int Soc Sports Nutr (2012) 9(1):45. doi:10.1186/1550-2783-9-45
CrossRef Full Text | Google Scholar
140. Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell (2013) 155(7):1451–63. doi:10.1016/j.cell.2013.11.024
PubMed Abstract | CrossRef Full Text | Google Scholar
141. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A (2010) 107(27):12204–9. doi:10.1073/pnas.0909122107
PubMed Abstract | CrossRef Full Text | Google Scholar
142. Telesford KM, Yan W, Ochoa-Reparaz J, Pant A, Kircher C, Christy MA, et al. A commensal symbiotic factor derived from Bacteroides fragilis promotes human CD39(+)Foxp3(+) T cells and Treg function. Gut Microbes (2015) 6(4):234–42. doi:10.1080/19490976.2015.1056973
PubMed Abstract | CrossRef Full Text | Google Scholar
143. Ochoa-Reparaz J, Mielcarz DW, Wang Y, Begum-Haque S, Dasgupta S, Kasper DL, et al. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol (2010) 3(5):487–95. doi:10.1038/mi.2010.29
PubMed Abstract | CrossRef Full Text | Google Scholar
 Derek low
Derek low